近日,图书馆VIP合肥微尺度物质科学国家研究中心和化学物理系曾杰教授研究团队设计构筑了铜-碳化铁界面型催化剂,实现了常压下二氧化碳加氢高选择性制备长链烯烃。相关成果以“Ambient-pressure hydrogenation of CO2 into long-chain olefins”为题发表在《自然·通讯》上,论文的共同第一作者是博士生李钟灵、特任副研究员吴文龙。
长链烯烃(C4+=)在精细化工领域具有广泛的应用,例如用于合成洗涤剂、高辛烷值汽油、润滑油、农药、增塑剂等。目前合成长链烯烃的主要途径是依赖于石油化工工业的烯烃聚合。如果利用可再生能源电解水制氢,再与温室气体二氧化碳反应直接制备长链烯烃,则会有巨大的环境效益。由于电解水设备规模小、布局分散,为了直接对接电解水制氢,需要使二氧化碳加氢反应在常压下进行。然而,目前二氧化碳加氢制备长链烯烃多在高压反应条件下进行。并且,根据勒夏特列原理,常压不利于长链烯烃的形成。因此,实现二氧化碳常压加氢制备长链烯烃仍然是一个巨大的挑战。
二氧化碳加氢制备烯烃主要通过甲醇中间体和一氧化碳中间体路径。由于低压既不利于甲醇合成反应也不利于甲醇制烃反应,研究团队选择了一氧化碳中间体路径。该路径的挑战在于在常压下设计合适的费托合成(FTS)活性位点。借鉴用于合成醇的改性费托催化剂的设计思路,研究人员在铁基催化剂的基础上引入具有一氧化碳非解离吸附能力的铜位点,制备了具有铜-碳化铁界面在常压下工作的铜-铁催化剂,该催化剂包含金属铜、四氧化三铁和碳化铁等多种物相。
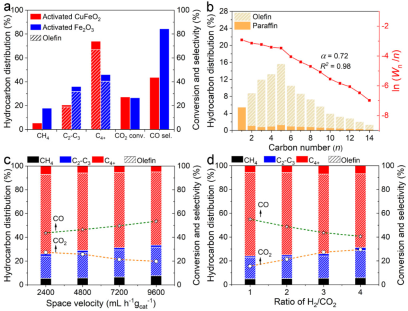
图1.(a)与传统铁基催化剂性能对比。(b)烃类产物碳数分布。(c)不同的空速下的性能对比。(d)不同H2/CO2比下的性能对比。
对催化剂进行性能评估发现在320oC,1 bar(H2:CO2 = 3:1),空速为2400 mL h-1 gcat-1的反应条件下,该催化剂对长链烯烃选择性高达66.9%,同时二氧化碳转化率为27.3%,一氧化碳选择性为43.7%。对比于传统铁基催化剂,该催化剂对一氧化碳和甲烷的选择性更低,对长链烯烃的选择性更高(图1a,b)。为了评价催化剂的适用性,我们改变了反应空速和H2/CO2的比值,当反应空速增大时,二氧化碳转化率降低但长链烯烃选择性未发生显著衰减,当H2/CO2的比值增大时长链烯烃选择性也仅轻微降低,这表明催化剂能适用于广泛的反应条件(图1c,d)。尽管催化剂在常压下操作,但其对长链烯烃的选择性与目前文献报道的高压反应条件下的最优值基本相当(图2)。研究人员发现,该催化剂经过长时间反应后长链烯烃选择性会下降,但经过简单的再生处理即可使其长链烯烃选择性恢复。

图2.(a)二氧化碳加氢制长链烯烃催化剂的选择性比较。
研究人员结合原位表征和理论计算研究了碳碳键的偶联机制。一氧化碳程序升温脱附实验证明了活化后的催化剂存在一氧化碳非解离吸附和解离吸附两种活性位点,说明可能存在一氧化碳插入机制(图3a)。同步辐射真空紫外光电离飞行时间质谱检测到微量含氧产物乙醛,也侧面印证了一氧化碳插入机制的存在(图3b)。理论计算的结果表明在铜-碳化铁界面处发生一氧化碳插入的反应能垒远低于单独的碳化铁,并且其CH2偶联的反应能垒也低于单独的碳化铁(图3c,d)。因此,研究人员认为,除了与传统铁基催化剂一样在碳化铁上通过碳化物路径进行碳碳偶联外,铜-碳化铁界面处还可以进行一氧化碳插入过程进行碳链增长,这样可以利用大量催化剂表面由于低压而未解离的一氧化碳。碳化物路径和一氧化碳插入路径的协同作用导致了良好的长链烯烃选择性。

图3.(a)一氧化碳程序升温脱附谱图。(b)光电离能谱图。(c)一氧化碳插入机制能垒。(d)碳化物能垒。
这项研究结果揭示了二氧化碳加氢反应过程中的碳碳键偶联机制,也为二氧化碳加氢制长链烯烃催化剂的设计提供了思路。该项研究得到了国家重点研发计划、国家科技攻关计划、国家杰出青年科学基金、安徽省联合基金重点项目等项目的支持。
论文链接:https://www.nature.com/articles/s41467-022-29971-5